Background
Every blood vessel in the body is lined with a specialized layer of polarized cells known as endothelium. An essential function of the endothelial monolayer is the regulation of barrier integrity, which prevents the leakage of plasma and proteins out of the circulation while still permitting the flux of nutrients and immune cells to target tissues.
In principle, permeability of the endothelial monolayer can reflect contributions from flux between endothelial cells (paracellular leakage) and through individual endothelial cells (transcellular leakage, or transcytosis). It is widely accepted that paracellular leakage predominates during inflammatory states such as sepsis and acute lung injury (the latter known clinically as the acute respiratory distress syndrome, ARDS). Accordingly, by far the majority of research on endothelial permeability has focused on this route of endothelial permeability: the methods of study are relatively straight-forward and there is obvious relevance to human disease. In contrast, the contribution of transcytosis to overall endothelial permeability is relatively obscure, particularly in the setting of inflammation. This is largely due to technical difficulties in distinguishing transcellular permeability from intercellular gaps, particularly in a dynamic and quantifiable way. In addition, endothelial cells grown in culture appear to lose the ability to perform transcytosis as they are passaged. Much of the initial work on transcytosis used electron microscopy of animal tissues, an expensive and often a mostly descriptive endeavour. Transcytosis by endothelial cells (at least in the apical to basal direction) was best described in early studies for the plasma protein albumin and is mediated by caveolae, small vesicles that bud off from the apical endothelial surface and release their cargo at the basal membrane. This process requires the protein caveolin-1 and the large GTPase dynamin; the latter is thought to mediate the scission of internalized caveolae from the apical plasmalemma.
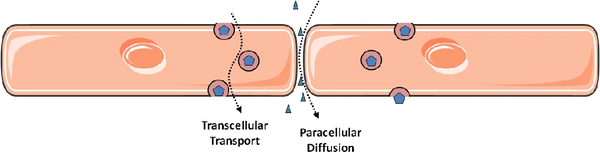
My lab is interested in both routes of endothelial permeability and how they are related:
We study paracellular leakage during inflammation, using acute lung injury induced by bacterial pathogens, the human influenza A virus or coronavirus as model systems. For example, we investigate how the influenza virus induces lung endothelial permeability to cause pulmonary edema, a characteristic clinical feature of severe influenza infections in humans. We have reported effects of the virus on lung endothelial viability and on tight junction integrity; interestingly, at least some of the effect of the virus on endothelial barrier integrity is independent of viral replication and involves degradation of the tight junction constituent claudin-5. Remarkably, restoration of endothelial barrier integrity using a Tie2 agonist peptide does not impair viral clearance and is sufficient to improve survival from otherwise fatal influenza in a pre-clinical model. Our findings add to the growing body of literature indicating that vascular leakage is not required for a competent innate immune response. Broadly speaking, this suggests that optimization of the host response to influenza (e.g. strengthening vascular integrity) represents a viable and unappreciated therapeutic approach. Unlike therapies that directly target the pathogen (e.g. antivirals, antibiotics), modulation of the host response is less likely to stimulate pathogen resistance. This - the development of host-modulating therapies for pathogen-induced lung injury - is now a major focus of the lab. We have developed methods for enhancing delivery of drugs and genes to the injured lung with the hope of inducing lung endothelial repair. We are also interested in the development of novel diagnostic approaches for lung endothelial permeability during pathogen-induced inflammation.

Another area of study in the lab is the contribution of endothelial transcytosis to the overall permeability of the endothelium to various macromolecules (e.g. insulin, albumin, lipoproteins).
Interestingly, endothelial transcytosis underlies the first stage of atherosclerosis. Accumulation of LDL-derived cholesterol under the arterial endothelium triggers an inflammatory reaction that culminates in luminal narrowing and eventually an unstable arterial plaque. However, how the LDL gets under the endothelium is poorly understood. Autopsy studies on young individuals dying of non-cardiac causes reveal a healthy, continuous endothelial layer overlying cholesterol deposits and the average LDL particle is too large to pass through intact cell-cell junctions; thus, LDL is likely to cross the endothelium by transcytosis. The canonical model of LDL receptor-initiated endocytosis does not explain LDL accumulation in the arterial intima.
We devised an assay to quantify LDL transcytosis by individual cells in a confluent monolayer using total internal reflection fluorescence microscopy (TIRF; see video below), complimented by traditional transwell assays and an ex vivo perfusion assay. The TIRF assay in particular is well suited to mechanistic studies and has numerous advantages over transwells and electron microscopy. Using this approach, we were the first to report an unexpected role for the scavenger receptor SR-BI in LDL transcytosis (Armstrong et al., 2015) - a finding since validated by others - and have shown how it is regulated by physiological concentrations of estrogen (Ghaffari et al., 2018). This latter effect may contribute to the lower rate of coronary artery disease observed in premenopausal women compared to age-matched men. Collaborative work led by the Sessa lab has established that blocking LDL transcytosis prevents atherosclerosis, implying that it is a veritable therapeutic target. Elucidating the regulation of transcytosis of LDL and other lipoproteins by coronary artery endothelial cells and how this affects atherosclerosis is now a major focus of the lab. For instance, we and others have reported that inflammation (a known risk factor for heart disease) enhances transcytosis of LDL.
Understanding how endothelial transcytosis of lipoproteins is regulated is likely to have broad-reaching pathologic and therapeutic implications.
The lab is located in the Keenan Research Centre for Biomedical Science, St. Michael's Hospital (a tertiary care teaching hospital) - part of the Unity Health Toronto network. Dr. Lee and the hospital are affiliated both with the University of Toronto and with Toronto Metropolitan University (formerly known as Ryerson).